Les cas les plus courants : le rétinoblastome unilatéral
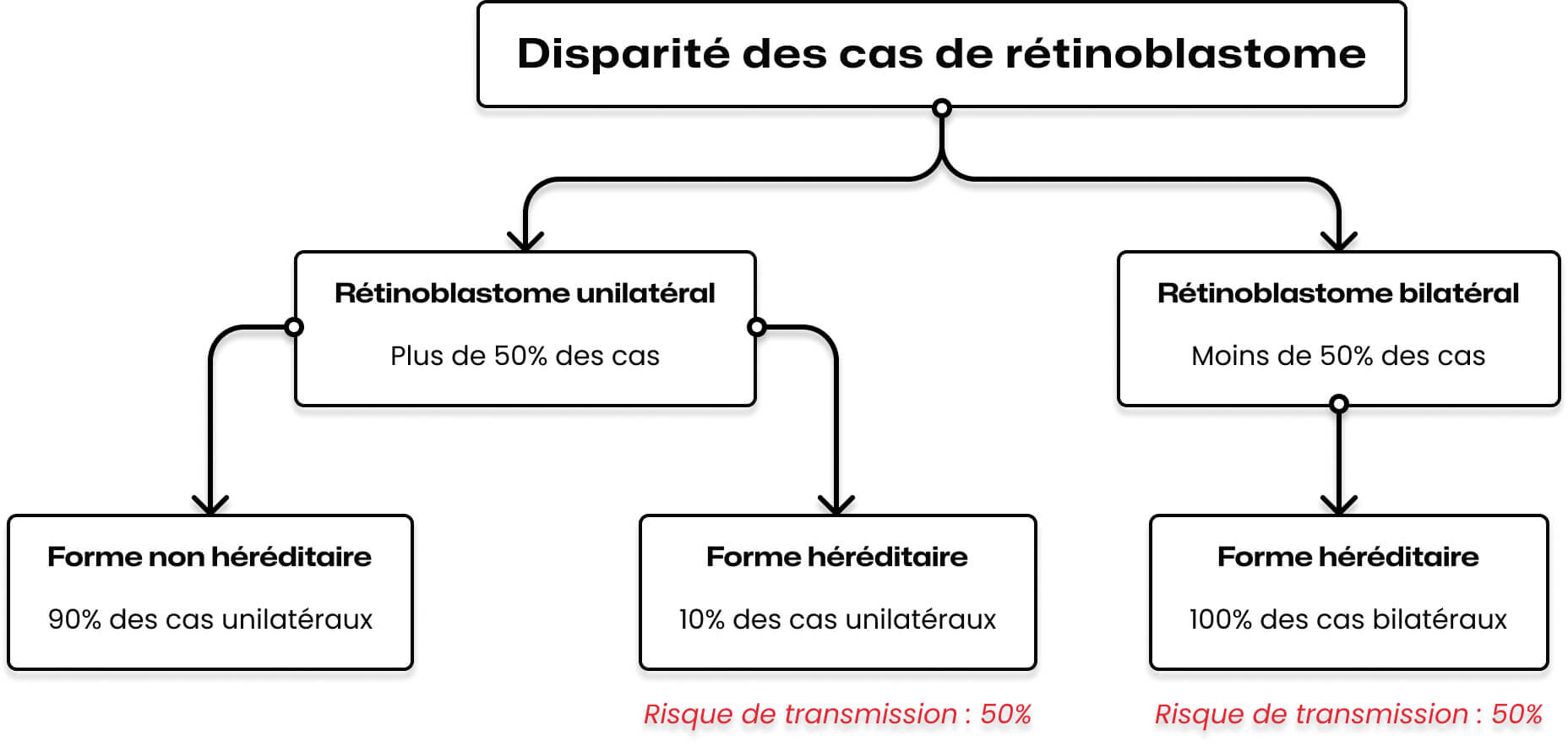
Dans plus de la moitié des cas de rétinoblastome, l’atteinte est unilatérale. Dans la très grande majorité de ces cas (90%), il s’agit de l’altération des deux exemplaires du gène RB1 au niveau d’une cellule de la rétine acquise au cours de la petite enfance.
Toutefois, il faut savoir que 10% des enfants atteints d’un rétinoblastome unilatéral sont porteurs d’une prédisposition génétique.
Dans 75% des cas, la maladie n’a aucune histoire familiale, il s’agit alors le plus souvent d’un accident génétique qui a eu lieu dans une cellule germinale (spermatozoïde ou ovocyte) de l’un des deux parents.
Dans ce cas, le risque de prédisposition des frères et sœurs de l’enfant atteint est très faible. En revanche, l’enfant porteur de cette prédisposition aura plus tard un risque sur deux de la transmettre à chacun de ses enfants, et un risque plus important que la population générale de développer un autre cancer (cancer de la peau, des os).
De ce fait, toute tuméfaction ou douleur survenant même des années après le traitement nécessitent d’être signalées au médecin traitant pour explorations complémentaires.
Pour tous les patients atteints d’un rétinoblastome, il existe donc un risque plus ou moins important d’être porteur d’une altération constitutionnelle du gène RB1, avec, alors, un risque de prédisposition pour la fratrie ou pour les descendants du patient. Un suivi précoce et fréquent, par examen du fond d’œil jusqu’à l’âge de 5 ans, est recommandé pour tout enfant ayant un risque de prédisposition au rétinoblastome.